Jak liczy komputer DNA
Obliczenia biomolekularne, biologia obliczeniowa, DNA komputery – to tylko niektóre ze stosowanych obecnie określeń na dynamicznie rozwijającą się dziedzinę wiedzy z pogranicza biologii molekularnej, inżynierii genetycznej i informatyki.
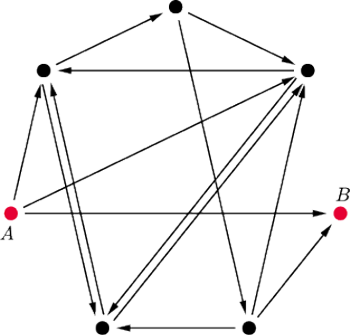
Rys. 1 Graf użyty w doświadczeniu Adlemana. Szukamy drogi Hamiltona z
 do
do
Rys. 1 Graf użyty w doświadczeniu Adlemana. Szukamy drogi Hamiltona z
 do
do

Trochę historii
Prawdziwe zainteresowanie świata nauki badaniami nad zastosowaniem reakcji
biomolekularnych w obliczeniach przyniosła praca Leonarda Adlemana z 1994
roku, w której zastosowano cząsteczki kwasu dezoksyrybonukleinowego (DNA)
oraz standardowe techniki inżynierii genetycznej w rozwiązaniu znanego w
matematyce problemu drogi Hamiltona na przykładzie 7 miast połączonych 14
drogami. Problem ten można sprowadzić do pytania: „Jak można
odwiedzić 7 miast połączonych 14 drogami, bez dwukrotnego przechodzenia
przez to samo miasto?”. Obliczenia, które były w istocie żmudnymi
laboratoryjnymi doświadczeniami, trwały tydzień, a warto dodać, że
człowiek rozwiązuje to samo zadanie z użyciem kartki i ołówka (można
także zastosować długopis) w czasie ledwie 1 minuty (!). Adleman wybrał nie
bez powodu zadanie szukania drogi Hamiltona, gdyż należy ono do tzw.
zadań NP-trudnych, czyli takich, dla których nie znamy odpowiednio
szybkich dokładnych algorytmów. Wszystkich możliwych cykli Hamiltona
dla n miast w grafie pełnym jest aż
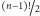 . Dla przykładu, dla 10
miast mamy 181 440 możliwych marszrut, dla 12 miast już prawie 20
milionów. Poszukiwania optymalnej drogi dla kilkudziesięciu miast
z zastosowaniem klasycznego komputera trwałyby wiele dziesiątków
lat.
. Dla przykładu, dla 10
miast mamy 181 440 możliwych marszrut, dla 12 miast już prawie 20
milionów. Poszukiwania optymalnej drogi dla kilkudziesięciu miast
z zastosowaniem klasycznego komputera trwałyby wiele dziesiątków
lat.
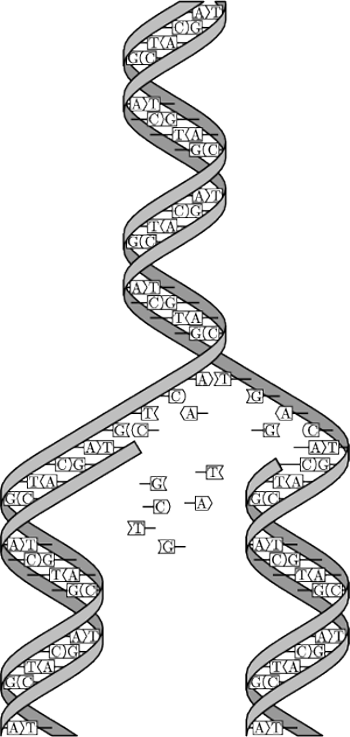
Rys. 2 Łańcuch DNA (Wikipedia).
Obecne komputery DNA osiągają prędkość reakcji molekularnych 330
TFLOPS (330 bilionów operacji zmiennoprzecinkowych na sekundę, czyli
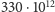 ) i to w objętości 5 mililitrów (objętości łyżeczki od
herbaty). Najszybszy komputer krzemowy, Blue Gene/L firmy IBM, osiągnął
w połowie ubiegłego roku prędkość 596 TFLOPS. Komputery DNA są jednak
nie tylko szybkie, ale charakteryzują się niezwykłą gęstością upakowania
informacji oraz znikomym zużyciem energii. Wystarczy powiedzieć, że to, co
obecnie wymaga zapisania na ponad bilionie CD-ROM-ów, zajęłoby około 1
cm
) i to w objętości 5 mililitrów (objętości łyżeczki od
herbaty). Najszybszy komputer krzemowy, Blue Gene/L firmy IBM, osiągnął
w połowie ubiegłego roku prędkość 596 TFLOPS. Komputery DNA są jednak
nie tylko szybkie, ale charakteryzują się niezwykłą gęstością upakowania
informacji oraz znikomym zużyciem energii. Wystarczy powiedzieć, że to, co
obecnie wymaga zapisania na ponad bilionie CD-ROM-ów, zajęłoby około 1
cm
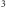 równoważnego 1 gramowi DNA, a 1 dżul pozwala na
wykonanie około
równoważnego 1 gramowi DNA, a 1 dżul pozwala na
wykonanie około
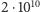 –krotnie więcej operacji w biokomputerze niż
w komputerze krzemowym. Dzisiaj obliczenia biomolekularne stosuje się,
między innymi, w rozwiązywaniu zadań NP-trudnych, budowie bardzo
dużych pamięci, masowych obliczeniach równoległych czy w konstrukcji
molekularnych układów elektronicznych.
–krotnie więcej operacji w biokomputerze niż
w komputerze krzemowym. Dzisiaj obliczenia biomolekularne stosuje się,
między innymi, w rozwiązywaniu zadań NP-trudnych, budowie bardzo
dużych pamięci, masowych obliczeniach równoległych czy w konstrukcji
molekularnych układów elektronicznych.
Trochę inżynierii genetycznej
No dobrze – mógłby teraz ktoś powiedzieć. Rzeczywiście wygląda to wszystko imponująco, ale tak naprawdę, jak to działa? Jak można, używając molekuł DNA, zrealizować jakiekolwiek obliczenia? W rzeczywistości komputer DNA opiera się na stosunkowo prostych mechanizmach genetyki molekularnej, a tak naprawdę cały problem leży, po pierwsze, w dobrym modelu obliczeń, a następnie prawidłowym doborze wszystkich parametrów laboratoryjnego eksperymentu. W obliczeniach biomolekularnych wszelkie sygnały koduje się za pomocą cząsteczek DNA. DNA jest polimerem składającym się z ciągu nukleotydów: adeniny (oznaczanej symbolem A), tyminy (T), cytozyny (C) oraz guaniny (G). Enzym polimerazy na podstawie jednej nici DNA potrafi stworzyć nić komplementarną, w której – zgodnie z zasadą komplementarności Watsona–Cricka – zamiast C pojawia się G, zamiast T – A, i na odwrót. Łańcuch DNA może być jednoniciowy lub dwuniciowy. Łańcuch dwuniciowy powstaje dzięki wiązaniom pomiędzy naprzeciwległymi, komplementarnymi nukleotydami (rys. 2). Dwie komplementarne nici DNA owijają się wokół wspólnej osi, tworząc tzw. podwójną helisę. Łańcuch dwuniciowy może mieć dwie orientacje, tzw. 3’ – 5’ oraz 5’ – 3’. Jeżeli dwa łańcuchy jednoniciowe są komplementarne i mają przeciwną orientację, to są lepkie (ang. sticky).
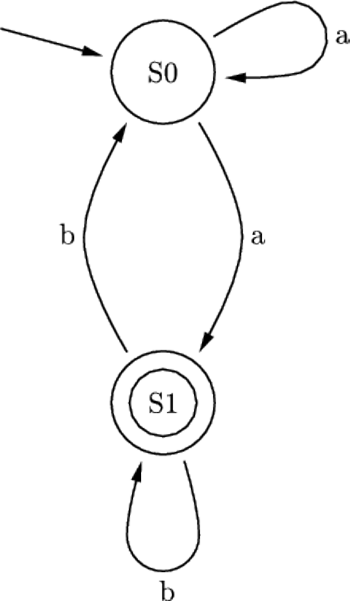
Rys. 3 Przykładowy automat skończony z dwoma stanami S0 i S1. Nieetykietowana strzałka
wskazuje na stan początkowy S0, podwójny okrąg oznacza stan końcowy S1. Etykietowane
symbolami alfabetu strzałki reprezentują możliwe przejścia automatu: S0
 aS0 (będąc
w stanie S0, po wczytaniu litery a, można przejść do stanu S0), S0
aS0 (będąc
w stanie S0, po wczytaniu litery a, można przejść do stanu S0), S0
 aS1
(ze stanu S0, po wczytaniu a, można przejść do S1), podobnie S1
aS1
(ze stanu S0, po wczytaniu a, można przejść do S1), podobnie S1
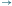 bS0, S1
bS0, S1
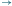 bS1.
bS1.
Na przykład, przetwarzając słowo abbab, automat mógłby przechodzić kolejno przez stany S0, S1, S1, S0, S1, S1.
Rys. 3 Przykładowy automat skończony z dwoma stanami S0 i S1.
W odpowiednich warunkach mogą one w procesie hybrydyzacji utworzyć łańcuch dwuniciowy, który stabilizuje się przez działanie enzymu ligazy. Istotnym narzędziem w inżynierii genetycznej jest endonukleaza restrykcyjna (restryktaza), która rozcina podwójną nić DNA. W przypadku restryktaz typu II cięcie następuje zawsze w określonym miejscu. Na przykład restryktaza FokI rozpoznaje w ciągu DNA sekwencję startową

i rozcina dwuniciowy łańcuch w sposób następujący

co zapisuje się
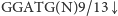 , gdzie
, gdzie
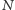 jest dowolnym
nukleotydem. Zatem cały mechanizm wykorzystywany w komputerze DNA
opiera się w istocie na odpowiednim rozcinaniu łańcucha dwuniciowego
i ponownym sklejaniu jego „lepkich” końców.
jest dowolnym
nukleotydem. Zatem cały mechanizm wykorzystywany w komputerze DNA
opiera się w istocie na odpowiednim rozcinaniu łańcucha dwuniciowego
i ponownym sklejaniu jego „lepkich” końców.
Automat molekularny Shapiry
W 2001 roku w prestiżowym czasopiśmie Nature ukazała się praca izraelskich naukowców z Instytutu Weizmanna w Rehovot opisująca molekularny komputer DNA. Dwa lata później ten sam zespół pod kierunkiem prof. Ehuda Shapiry zaprezentował ulepszoną wersję molekularnej maszyny, która nie tylko była w tym czasie najszybszym komputerem na świecie, ale również i najmniejszym. Co więcej, ów komputer praktycznie nie potrzebuje żadnej energii zewnętrznej dla swego działania, gdyż dostarczają jej same cząsteczki DNA biorące udział w obliczeniach.
Komputer prof. Shapiry jest przykładem molekularnej realizacji automatu
skończonego. Automat skończony jest pewną podklasą tzw. maszyny
Turinga, która jest z kolei abstrakcyjnym modelem dowolnego komputera.
Automat skończony operuje na wejściowej sekwencji symboli. Automat
może znajdować się w jednym z ustalonej liczby stanów wewnętrznych
(reprezentujących pamięć komputera), z których jedne są traktowane jako
stany początkowe, a inne jako stany końcowe. Oprogramowanie automatu
stanowi zbiór reguł przejść, z których każda określa sposób,
w jaki mają zmieniać się stany automatu pod wpływem pojawiających się
symboli wejściowych i aktualnego stanu maszyny. Automat może nie
kończyć obliczeń, jeżeli żadna z dostępnych reguł nie może być
zastosowana. Obliczenia natomiast kończą się, gdy przetworzony zostanie
ostatni symbol wejściowy. Automat akceptuje dane wejściowe, jeżeli
obliczenia zatrzymują się w stanie końcowym automatu. Rysunek 3 przedstawia
przykład automatu dwustanowego, pracującego na dwuliterowym alfabecie
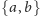 .
.
Automat Shapiry jest właśnie molekularną implementacją automatu złożonego z 2 stanów i operującego na 2 literach. Można by zapytać, czy tak prosty model może mieć jakieś praktyczne zastosowanie? Jeżeli weźmiemy pod uwagę fakt, że w takim automacie można zdefiniować 255 różnych zestawów przejść, to w połączeniu z 3 różnymi konfiguracjami stanów końcowych (S0 lub S1, lub S0 i S1), daje w rezultacie możliwość implementacji 765 programów. A to już jest niemało. W modelu Shapiry założono, że każdy symbol wejściowy kodowany jest na 6 parach nukleotydów (szczegóły na marginesie). Molekuła wejściowa określa stan początkowy automatu oraz sekwencję wejściową. Architekturę danego automatu tworzy zestaw molekuł reprezentujących przejścia automatu, wybrany z grupy 8 możliwych przejść automatu dwustanowego. Dodatkowo dostępne są dwie molekuły wyjściowe, zadaniem których jest rozpoznawanie osiągnięcia przez automat stanu końcowego. Kodowanie molekuł uwzględnia miejsce cięcia restryktazy FokI.
Obliczenia rozpoczynają się w momencie zmieszania molekuły wejściowej, molekuł reprezentujących przejścia automatu oraz restryktazy FokI. Przedstawimy przykładowe działanie automatu z rysunku 3 akceptującego słowo wejściowe abbab. Oto jak zakodowany zostaje stan początkowy i ciąg wejściowy: (Dane wejściowe. Kolorem zaznaczono miejsca, w których zakodowane są dwie litery a ze słowa abbab.
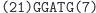
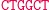
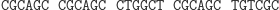
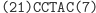
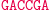
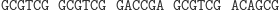
W pierwszym kroku restryktaza rozcina molekułę wejściową, eksponując 4-nukleotydowy lepki koniec kodujący stan początkowy i pierwszy symbol wejściowy.
Działanie restryktazy FokI:
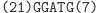

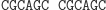
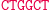
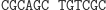
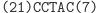
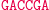
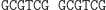
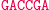
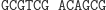
W następnym kroku do lepkiego końca molekuły wejściowej przyłącza się poprzez działanie ligazy pasujący (komplementarny) koniec jednej z molekuł przejścia automatu.
Ligacja z udziałem reguły S0
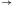 aS1:
aS1:
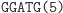
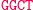
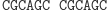
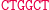
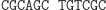
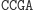
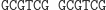
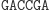
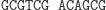
Ta hybrydyzacja oznacza przejście automatu do kolejnego stanu i wczytanie następnego symbolu wejściowego. W kolejnym kroku produkt hybrydyzacji jest znowu rozcinany przez FokI i tworzony jest lepki koniec reprezentujący kolejny stan i symbol wejściowy. Cały proces przebiega w pętli do momentu połączenia lepkiego końca z molekułą wyjściową, reprezentującą stan końcowy:
Działanie restryktazy FokI:
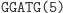
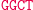
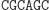
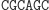
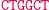
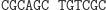
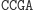
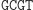
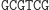
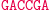
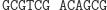
Ligacja z udziałem reguły S1
 bS0:
bS0:
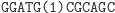
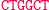
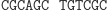
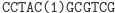
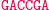
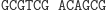
Działanie
restryktazy FokI:
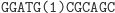

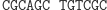
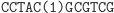
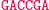
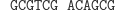
Ligacja
z udziałem reguły S0
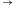 aS1:
aS1:
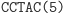
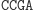
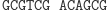
Działanie
restryktazy FokI:
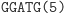
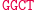
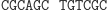
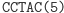
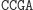
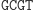
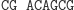
Ligacja
z udziałem reguły S1
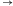 bS1:
bS1:
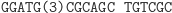
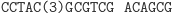
Działanie restryktazy FokI:
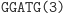
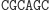
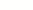
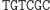
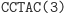
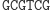
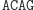

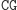
Ligacja
z udziałem reguły detektora S1-D:
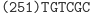
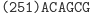
Zamiast zakończenia
Wizja komputera pracującego z olbrzymią prędkością, praktycznie bez zewnętrznej energii, o niewiarygodnym wręcz stopniu upakowania informacji zachęca do coraz to nowych poszukiwań. W kwietniowym wydaniu Nature z 2004 r. ukazał się artykuł, w którym prof. Ehud Shapiro opisał zastosowanie komputera DNA do analizy biologicznych informacji przechowywanych w mRNA. W warunkach laboratoryjnych biokomputer zdiagnozował typ komórek rakowych oraz rozpoczął proces zwalczania ich przez produkcję odpowiednich substancji. Również w 2004 r. piszący te słowa wraz z doktorantem Maciejem Trociem zaproponowali zastosowanie nowych enzymów restrykcyjnych w konstrukcji molekularnego automatu, a także zamodelowali działanie automatu 3-stanowego, zdolnego wykonywać już 1 835 001 programów. W innej publikacji wskazali na możliwość realizacji biologicznych funkcji logicznych oraz systemu wnioskowania, który może znaleźć zastosowanie np. w molekularnym systemie ekspertowym. W 2005 r. zespół izraelskich naukowców opublikował wyniki realizacji automatu 3-stanowego, nad alfabetem 3-literowym. Praca wskazywała również na teoretyczne szanse budowy komputera rozpoznającego 39 różnych symboli wejściowych. W ubiegłym roku ukazała się praca Chińczyków, którzy eksperymentalnie udowodnili, że można konstruować komputery DNA wykorzystujące restryktazy z tzw. grupy IIS bez użycia ligazy. W sposób istotny może to uprościć przyszłą realizację biokomputerów.